A clinical hold from the U.S. Food and Drug Administration can significantly prolong the time and increase the cost of drug development, which is particularly concerning for emerging/small biotech and specialty pharma companies. In this blog, we discuss common reasons for clinical holds and provide useful tips for both avoiding and addressing them.
Brief background on clinical holds
Following the submission of an investigational new drug (IND) application, the FDA has 30 days to review the application to ensure study participants will not be subjected to unreasonable risk and the study is designed to address its stated objectives. The FDA may issue a clinical hold if there are questions about critical aspects of clinical study design and safety, non-clinical safety, or product quality of the proposed investigational treatment that are not readily resolvable, or there is an inability to conduct the clinical study in accordance with good clinical practice (GCP) guidelines and industry standards.
If a proposed clinical investigation is put on a clinical hold, the sponsor is not permitted to give the investigational treatment to subjects. If an ongoing study is placed on a clinical hold, no new subjects may be administered the investigational therapy. Typically, patients already in the study must be taken off the investigational treatment. Still, there are cases where the FDA permits ongoing administration if the potential benefit appears to outweigh the risks of continued treatment.[1]
Common reasons for clinical holds
In recent months, the FDA has placed clinical holds on a number of gene therapy, oncology, and rare disease trials for reasons ranging from the need for additional safety data to concerns about product quality; ability to measure clinical benefit, especially in high-risk pediatric patient populations; and justification for starting dose and dose escalation in a clinical Phase 1 first-in-human study.
According to FDA research, the most common deficiencies leading to clinical holds were product quality issues, followed by clinical and toxicology concerns.[2] With gene therapies and other next-generation therapies, in particular, the manufacturing process often raises concerns due to impurities, degradation products, or metabolites that have not been qualified in a non-clinical safety model. In such cases, sponsors may need to perform additional studies or even change their manufacturing process to remove the clinical hold.
Five tips for avoiding a clinical hold
When preparing an IND, sponsors should focus on providing enough background information for the FDA to reasonably assess the risks to the subjects (see Figure 1). The key to avoiding a clinical hold is demonstrating the adequacy of the clinical protocol to address the stated goals and exercising due diligence to ensure the proposed study adequately controls risk.
Figure 1. Product development framework for IND submission
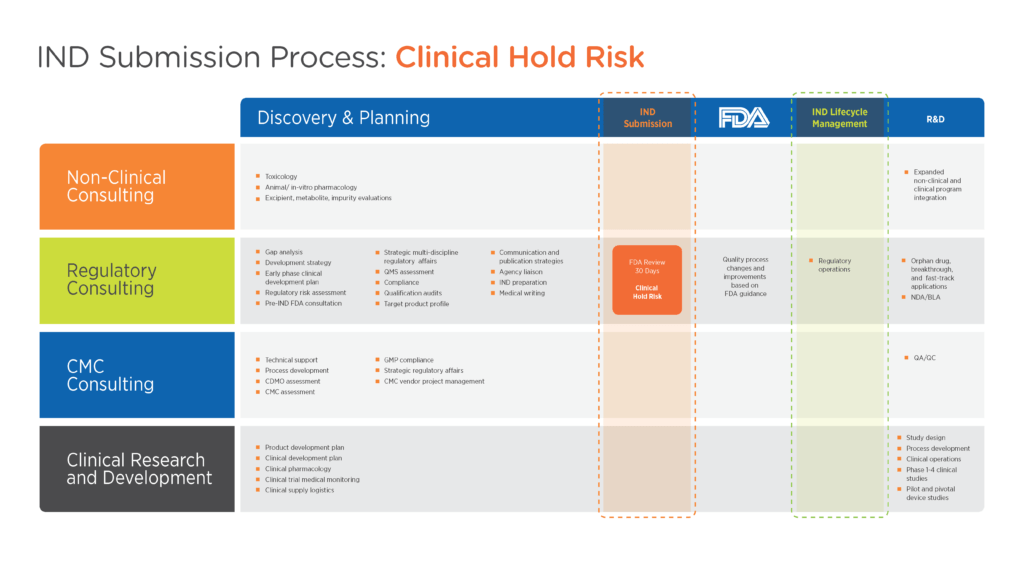
Click image to enlarge
- Select the right toxicology animal model.
Non-clinical safety studies need to be sufficiently comprehensive for evaluation of the investigational treatment in humans. In particular, non-clinical toxicology studies are important, and the duration of those studies – especially repeat dose – will depend on the proposed treatment duration in Phase 1. - Document and validate the manufacturing process.
It is critical to demonstrate that manufacturing will not introduce expected risks, including impurities or structurally similar materials that may be known to be carcinogenic or with structural alert concerns. - Document reasonable expectations of safety in the ICF.
While the FDA does not routinely require an informed consent form (ICF) as part of every IND, the agency may request one for certain high-risk study populations or therapeutic areas, such as pediatrics, congestive heart failure, and oncology. In these scenarios, failure to warn can be a serious issue that puts a sponsor at legal liability. Having an ICF ready at the time of IND submission eliminates the urgency in preparing one if unexpectedly requested during the FDA’s review. - Engage the right experts to understand all the potential risks.
Often, emerging biotech and specialty pharma companies are not fully staffed with subject matter experts in all the disciplines – pharmacology, toxicology, manufacturing, regulatory, and clinical – required to complete a robust IND. Engaging with a regulatory consultant who has the cross-functional depth and breadth of expertise needed to understand what limitations are likely to raise red flags for the FDA can be extremely valuable. - Don’t underestimate the importance of well-written documents.
The challenge of IND preparation is using the right data to tell the right story. Experienced medical writers are adept at organizing information and highlighting key messaging so that the story is conveyed in a way that is meaningful and consistent to the FDA reviewer.
Asking the right questions to address a clinical hold
Sponsors who receive notice of a clinical hold from the FDA should be prepared to communicate proactively with the agency to get answers to the following questions:
- Is the FDA placing a full or partial clinical hold?
With a partial hold, some trial investigations are allowed to continue. Clarifying whether dosing can continue without interruption in already-enrolled patients may be critical to preventing disruptions in data collection. - What is the nature of the FDA’s concern?
A clinical hold may be placed for clinical concerns, common concerns about chemistry, manufacturing, and controls (CMC), or issues regarding the management and control of the clinical study itself. It is important to distinguish the nature of the concern, as well as the detailed listing of reasons behind the clinical hold, in order to plan an appropriate resolution strategy. The FDA will tell you why the clinical hold is being placed and what needs to be done to get it lifted. Look closely at the language and respect the advice that the FDA provides. - Who needs to be notified?
Sponsors will need a communications strategy to communicate with sites, partners, institutional review boards and/or ethics committees, and other interests that may be contractually obligated to be notified of a clinical hold.[3] If the study is being conducted in countries other than the U.S., these health authorities should also be notified of the clinical hold (or not) in accordance with their regulations and guidelines. Public companies may be required to disclose a clinical hold to investors. Keep in mind that sponsors have the option of withdrawing an IND without penalty, readdressing the content, and resubmitting to the FDA.
The bottom line
It is incumbent upon sponsors to exercise due diligence in doing the work necessary to support an IND. Experienced regulatory consultants can provide the oversight necessary to help ensure that the full range of risks is identified, from risks that may exist for patients to those that can impact corporate reputation.
Premier Consulting offers full-service strategic and tactical product development expertise in all stages of drug development. Our regulatory consultants are with you throughout the development of your drug, biologic, device, drug-device combination, or diagnostic. For a personalized consultation with one of our experts on how to optimize your regulatory strategy, contact us.
[1] U.S. Food and Drug Administration. CFR – Code of Federal Regulations Title 21. Available at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=312.42. Accessed August 24, 2020.
[2] U.S. Food and Drug Administration. How do clinical holds impact drug development programs. Available at https://www.fda.gov/drugs/news-events-human-drugs/how-do-clinical-holds-impact-drug-development-programs. Accessed August 24, 2020.
[3] Goodwin. Clinical holds: Tips for handling FDA’s call and what to do next. Available at https://www.goodwinlaw.com/publications/2020/02/02_27-clinical-holds-tips-for-handling-fda-call. Accessed August 24, 2020.