美国食品和药物管理局的临床搁置会大大延长药物开发的时间并增加成本,这对新兴/小型生物技术公司和专业制药公司来说尤为重要。在本博客中,我们将讨论临床搁置的常见原因,并提供避免和解决这些问题的实用技巧。
临床搁置的背景简介
在提交研究性新药(IND)申请后,FDA 有 30 天的时间对申请进行审查,以确保研究参与者不会遭受不合理的风险,而且研究的设计是为了实现其既定目标。如果在临床研究设计和安全性、非临床安全性或拟议研究疗法的产品质量等关键方面存在问题且无法立即解决,或者无法按照良好临床实践 (GCP) 指南和行业标准开展临床研究,FDA 可能会发出临床搁置令。
如果一项拟议的临床研究被临床搁置,申办者不得向受试者提供研究性治疗。如果一项正在进行的研究被临床搁置,则不得向新的受试者提供研究性治疗。通常情况下,已在研究中的患者必须停止接受试验性治疗。不过,在某些情况下,如果潜在的益处似乎大于继续治疗的风险,食品及药物管理局还是允许继续给药的。
临床搁置的常见原因
近几个月来,美国食品及药物管理局对一些基因治疗、肿瘤和罕见病试验进行了临床搁置,原因包括需要更多的安全性数据、对产品质量的担忧、衡量临床获益的能力(尤其是在高风险儿科患者群体中)以及临床 1 期首次人体试验中起始剂量和剂量升级的合理性。
根据 FDA 的研究,导致临床搁置的最常见缺陷是产品质量问题,其次是临床和毒理学问题。[2]尤其是基因疗法和其他下一代疗法,其生产工艺往往会因杂质、降解产物或代谢物等未经非临床安全模型验证的问题而引起关注。在这种情况下,申办者可能需要进行额外的研究,甚至改变生产工艺,以取消临床保留。
避免临床搁置的五条建议
在准备 IND 时,申办者应重点提供足够的背景信息,以便 FDA 合理评估受试者面临的风险(见图 1)。避免临床搁置的关键在于证明临床方案足以实现既定目标,并尽职尽责地确保拟议研究能充分控制风险。
图 1.提交 IND 的产品开发框架
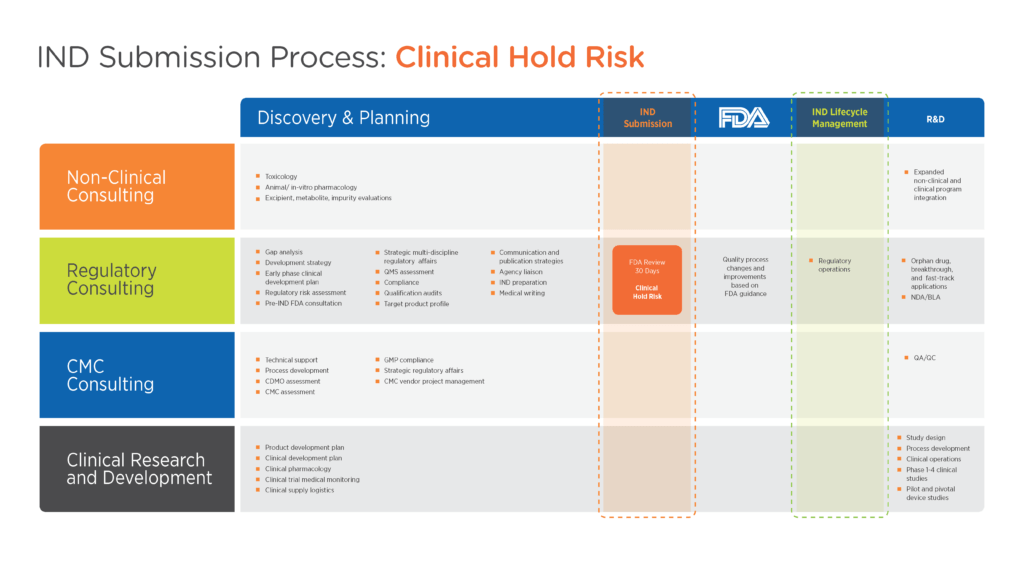
点击图片放大
- 选择合适的毒理学动物模型。
非临床安全性研究必须足够全面,以便对人体进行研究性治疗评估。非临床毒理学研究尤其重要,这些研究的持续时间(尤其是重复剂量)将取决于第一阶段的拟议治疗时间。
关键是要证明生产过程不会带来预期的风险,包括杂质或结构类似的材料,而这些材料可能是已知的致癌物质或具有结构警报问题。- 在 ICF 中记录对安全性的合理预期。
虽然 FDA 并不经常要求将知情同意书 (ICF) 作为每个 IND 的一部分,但对于某些高风险研究人群或治疗领域,如儿科、充血性心力衰竭和肿瘤科,该机构可能会要求提供知情同意书。在这些情况下,未发出警告可能是一个严重的问题,会使申办者承担法律责任。在提交 IND 时就准备好 ICF,就不会在 FDA 审查期间意外提出要求时急于准备 ICF。 - 聘请合适的专家了解所有潜在风险。
通常情况下,新兴生物技术和专业制药公司并不完全拥有完成稳健 IND 所需的药理学、毒理学、生产、监管和临床等所有学科的主题专家。如果监管顾问具有跨职能的深度和广度,能够了解哪些限制可能会引起 FDA 的警惕,那么聘请这样的监管顾问将非常有价值。
IND 准备工作的挑战在于使用正确的数据讲述正确的故事。经验丰富的医学撰稿人善于组织信息并突出关键信息,从而以对 FDA 评审员有意义且一致的方式传达故事。
提出正确的问题以解决临床搁置问题
收到 FDA 临床搁置通知的申办者应做好准备,积极与该机构沟通,以获得以下问题的答案:
在部分搁置的情况下,允许继续进行一些试验研究。明确已经入组的患者是否可以不间断地继续用药可能对防止数据收集中断至关重要。- FDA 所关注问题的性质是什么?
临床搁置可能是因为临床问题、有关化学、生产和控制 (CMC) 的常见问题或有关临床研究本身的管理和控制问题。重要的是要分清问题的性质,并详细列出临床搁置背后的原因,以便制定适当的解决策略。FDA 会告诉您临床搁置的原因,以及解除临床搁置需要采取的措施。请仔细阅读并尊重 FDA 提供的建议。
申办者需要制定沟通策略,与研究机构、合作伙伴、机构审查委员会和/或伦理委员会以及其他可能有合同义务被告知临床搁置的利益相关方进行沟通。[3]如果研究是在美国以外的国家进行,则还应根据这些国家的法规和指南将临床搁置(或不临床搁置)通知这些国家的卫生当局。上市公司可能需要向投资者披露临床搁置。请记住,申办者可以选择撤回 IND 而不受处罚,重新修改内容并重新提交给 FDA。
底线
申办者有责任尽职尽责地开展必要的工作,以支持 IND。经验丰富的监管顾问可以提供必要的监督,帮助确保识别各种风险,从可能存在的患者风险到可能影响企业声誉的风险。
Premier Consulting在药物开发的各个阶段提供全方位的战略和战术产品开发专业服务。我们的法规顾问在您的药物、生物制剂、器械、药物-器械组合或诊断产品的整个开发过程中始终与您在一起。如需与我们的专家就如何优化您的监管策略进行个性化咨询,请联系我们。
[1]美国食品和药物管理局。CFR - 《联邦法规汇编》第 21 篇。见https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=312.42。访问日期:2020 年 8 月 24 日。
[2]美国食品和药物管理局。临床试验如何影响药物开发计划。见https://www.fda.gov/drugs/news-events-human-drugs/how-do-clinical-holds-impact-drug-development-programs。2020年8月24日访问。
[3]古德温。临床搁置:处理 FDA 电话的提示及下一步行动。见https://www.goodwinlaw.com/publications/2020/02/02_27-clinical-holds-tips-for-handling-fda-call。2020年8月24日访问。