Rare cancers account for almost a quarter of all new cancers worldwide though there is no universally adopted definition for rare cancers. In the U.S., rare cancers are defined as those with fewer than 15 cases per 100,000 per year, whereas in the EU, they are defined as six cases per 100,000 per year.
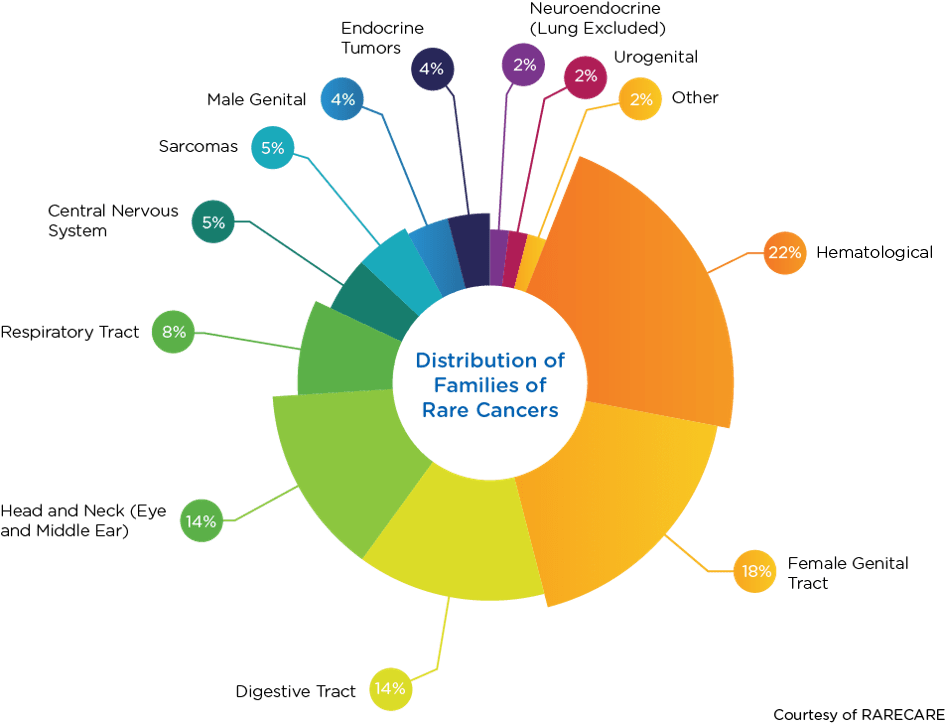
Historically, cancers have been characterized by location and tissue type. Today, however, cancers are increasingly being grouped based on molecular subtypes. This molecular approach to characterization has resulted in the categorization of some common cancers into groups of rare cancers. For example, melanoma is not a rare cancer, but when it is divided into molecular subtypes, it can be categorized as a collection of rare cancers. When grouped as families, the top four rare cancer types are hematological (22 percent), female genital tract (18 percent), digestive tract (14 percent), and head and neck (14 percent).
The grouping of cancer-based on molecular subtypes has changed not only how cancer is categorized but also how novel therapies are investigated in clinical trials. In this blog post, we explore the changing rare oncology landscape and novel approaches to rare cancer trials.
How rare cancers differ from rare diseases
Rare cancers are sometimes called the rare diseases of oncology. Yet, while rare cancers do share some similarities with rare diseases — namely small populations, geographic dispersion of patients, difficulties with diagnosis, and statistical challenges stemming from small sample sizes — there are some key differences:
- Definition – Rare diseases are defined based on prevalence, whereas rare cancers are defined based on incidence
- Genetic basis – 80 percent of rare diseases are of genetic origin, while few rare cancers have a well-defined genetic component
- Pediatric patients – 75 percent of rare diseases affect children, but all pediatric cancers are considered rare
- Target discovery – Rare diseases are a heterogeneous collection of diseases, whereas rare cancers all fall under the larger cancer umbrella and may benefit from discoveries in common cancers
The transition to precision medicine
Traditionally, cancer has been treated by surgery, radiation, and chemotherapy. Advances in genetics and molecular analysis techniques have made more customized patient care possible. Now, with precision or personalized medicine, treatments can be targeted to a subgroup of patients rather than relying on a one-drug-fits-all model.
As our understanding of the molecular characteristics of tumors has improved, there has been a tremendous leap forward —not only in targeted therapies but also in the development of tumor-or tissue-agnostic treatments. These therapeutics are based not on tumor location or tissue type but on molecular characterization. To date, three tumor agnostic therapies have received regulatory approval:
- Pembrolizumabin 2017 for tumors with microsatellite instability high (MSI-H) or deficient mismatch repair (dMMR)
- Larotrectinib in 2018 for tumors with NTRK gene fusions, which are found in approximately 0.3 percent of patients with varying cancer histologies
- Entrectinib in 2019 for tumors with NTRK gene fusions
Larotrectinib represents an important example of biomarker-driven, innovative drug development. In fact, it is the first drug fully developed using an agnostic indication approach. Approval of Larotrectinib was based on efficacy data from three individual open-label, single-arm trials. These trials were basket studies that included patients with 12 different solid tumor indications ranging from salivary gland cancer to infantile fibrosarcoma, all with the same NTRK gene fusion. All patients were treated with larotrectinib with an overall response rate of 75 percent. Even more impressively, approval was granted based on pulled data from only 55 patients across all 12 indications, and the timeframe from Phase 1 to accelerated approval was only three years.
Pushing the envelope with seamless oncology drug development
In oncology, particularly rare oncology, there are an increasing number of programs that have transitioned from a traditional drug development pathway to a seamless one, where pharmacology, therapeutic exploratory, and confirmatory studies are combined to pursue accelerated approval (see Figure 2).
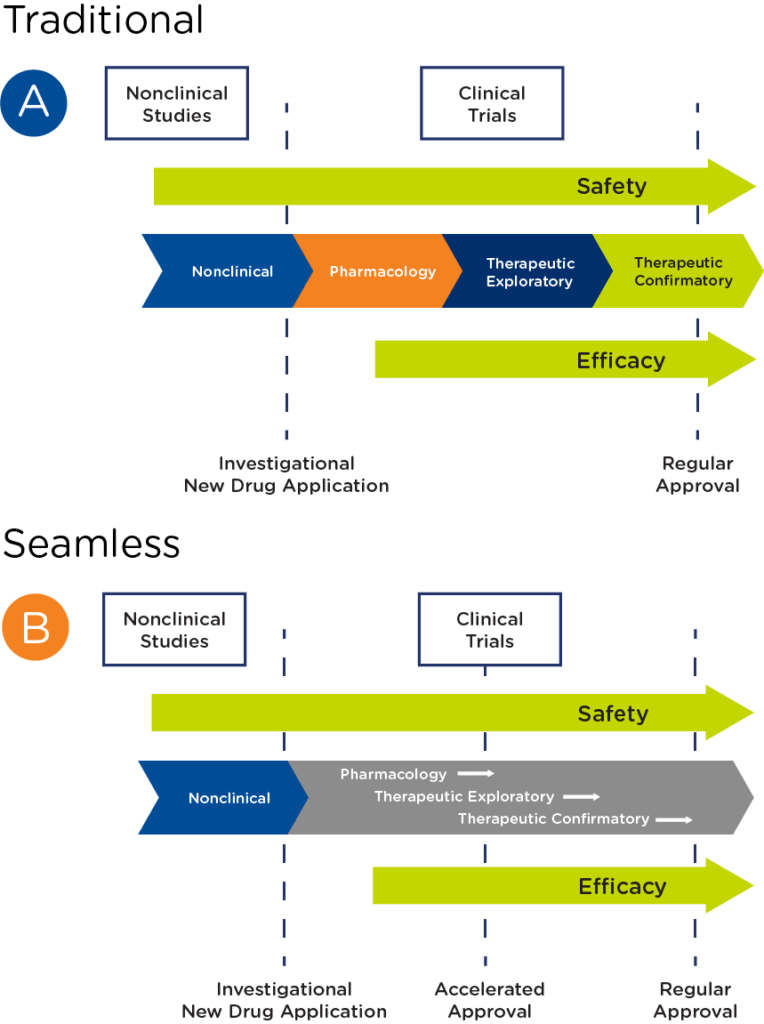
Challenges of a seamless approach to drug development in rare oncology include:
- Small populations – Given the rarity of the indication, the number of patients in the final dataset will be low
- Access to patients – To reach the patient number needed, a study will need to have a broad geographical reach, which increases logistical complexity
- Study design considerations – When preparing to design a rare oncology study, you will need to clearly define the trial objective, the study population and subgroups, and the methodology used to assess response. When defining the study population, it is important to consider how to handle patients who progress after initial treatment, especially if overall survival is an endpoint. With regard to response assessment, biomarkers may be more sensitive than tumor measurements in gauging response, particularly for targeted therapies and immunotherapies
- Incorporation of genetic or biomarker testing – You will need to select the right testing kit and testing methodology early in the program, ensuring it is fit for purpose based on the development stage but can also support further development and, ultimately, commercialization
- Manufacturing – With an accelerated program, manufacturing will need to be able to meet the requirements for a marketing application more quickly
Key takeaways
With shortened timelines, it is also critical to achieve alignment across the clinical, manufacturing, regulatory, and marketing disciplines on how to get to approval. Premier Research offers a full spectrum of product development consulting and clinical development services to help you optimize your rare oncology drug development. To schedule a conversation with one of our experts, click here.
1 Based on data from RARECARENet. Available at www.rarecare.eu.
2 Adapted from Theoret MR, Pai-Scherf LH, Chuk MK, et al. Clin Cancer Res October 15 2015 (21) (20) 4545-4551; DOI: 10.1158/1078-0432.CCR-14-3244