虽然医疗器械临床研究和试验药物临床研究有其不同之处,但它们的共同目标是保障研究参与者的权利和福利,同时尽可能快速有效地将安全有效的产品推向市场。
并非所有医疗器械都需要进行临床试验,这取决于其风险分层。但在需要进行临床试验时,器械申办者需要遵循许多与药品试验相同的监管要求,包括:
- 21 CFR 第 11 部分--电子记录;电子签名,其中规定了 FDA 认为电子记录、电子签名和在电子记录上执行的手写签名可信、可靠并与纸质记录和在纸质记录上执行的手写签名基本等同的标准。
- 美国联邦法规汇编》第 21 卷第 50 部分--《人类受试者保护》,涉及临床研究中儿童的知情同意和额外保障措施
- 21 CFR 第 54 部分--临床研究者的财务披露,要求申请人披露申办者与临床研究者之间的某些财务安排,以及临床研究者在所研究产品或申办者中的某些利益。
- 联邦法规汇编》第 21 卷第 56 部分--《机构审查委员会》,其中载有关于机构审查委员会的组成、运作和职责的一般标准,机构审查委员会负责审查临床调查,以支持美国食品及药物管理局监管产品(包括人用药品和医疗器械)的研究或营销许可申请。
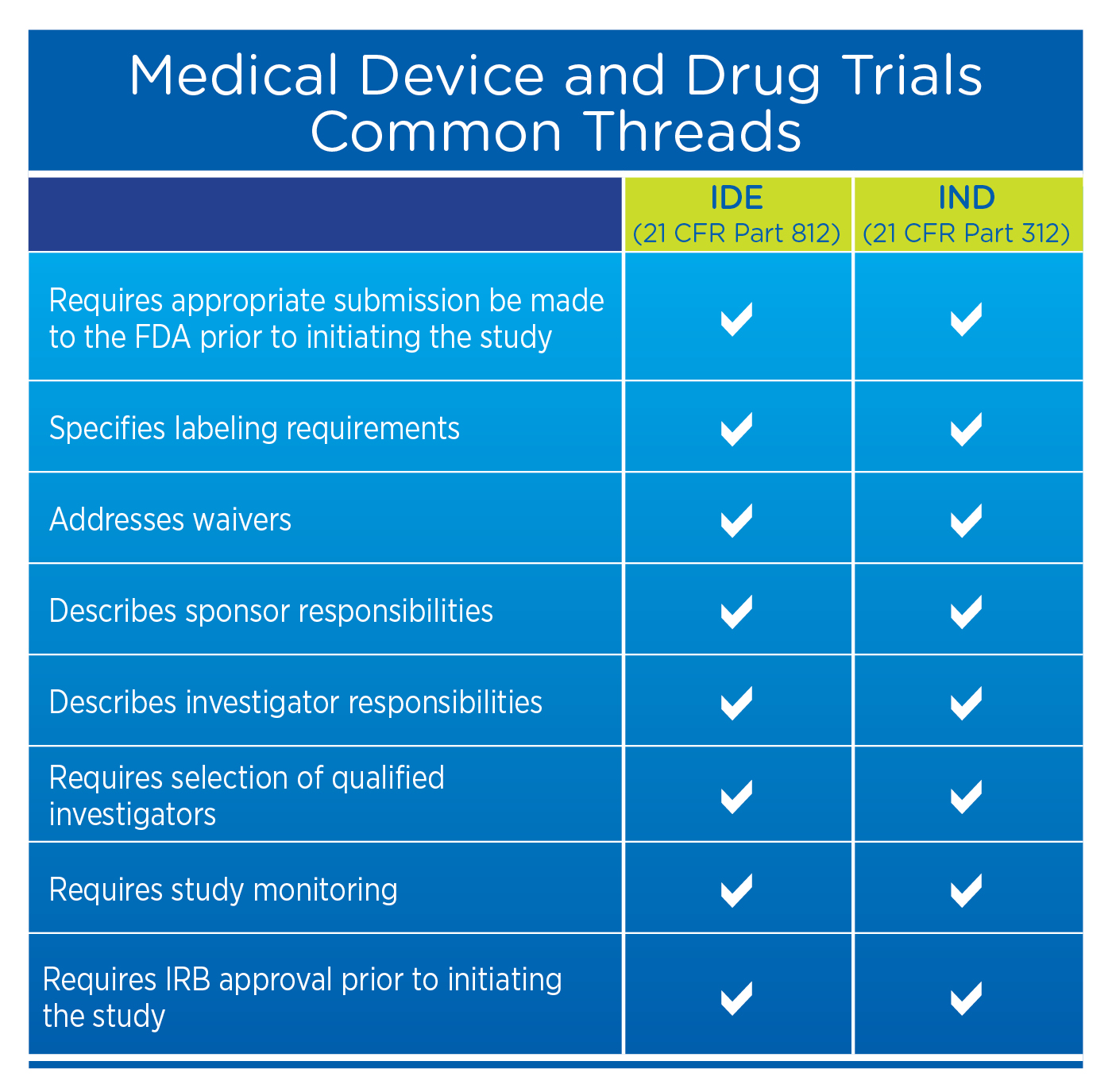
虽然器械申办者不需要完成新药研究申请(IND,根据《联邦法规汇编》第 21 卷第 312 部分),但他们需要完成器械研究豁免(IDE,根据《联邦法规汇编》第 21 卷第 812 部分),其中有许多类似的要求,如下表所示。
联邦对设备和药品的监管要求有何相似之处
然而,医疗器械试验与药物试验之间存在重大差异,器械公司在寻求将临床试验外包给合同研究组织(CRO)时,应确保合同研究组织拥有相关经验和适当资质的员工来处理所有要求。