在全球多中心研究变得越来越普遍的临床试验环境中,试验监测变得更加复杂。法规要求医疗器械研究的赞助者进行现场监测,以确保调查人员符合联邦法规、赞助者协议、调查计划以及调查审查委员会(IRB)批准的研究要求。此外,赞助商负责选择合格的监督员来监督试验参与者的安全。
申办者可以任命其他个人和团体,如数据监测委员会(DMC)或临床终点委员会(CEC),以确保合规性和适当的临床试验监测。但是,DMC和CEC之间有什么区别,申办者如何知道需要何种类型的研究监督?
数据监测委员会
FDA已经为临床试验申办者发布了关于建立和运行临床试验数据监测委员会的指南,该委员会也被称为数据和安全监测委员会(DSMBs)、数据和安全监测委员会(DSMCs),或独立数据监测委员会(IDMCs)。[1]该指南旨在帮助申办者确定DMC何时可用于研究监测以及此类委员会应如何运作。
FDA建议赞助商在以下情况下考虑使用DMC:1
- 研究终点是这样的:如果在中期分析中出现非常有利或不利的结果,包括发现无用的结果,可能在伦理上需要提前终止研究
- 有先验的安全考虑的理由,例如,一个特别具有侵入性的程序
- 之前有资料表明可能存在严重的毒性
- 该研究是在一个潜在的脆弱或易受伤害的人群中进行的。
- 该研究是在死亡或其他严重后果风险较高的人群中进行的。
- 研究规模大,或持续时间长,并且是多中心的。
DMC是一个由具有相关专业知识的个人组成的团体,他们。
- 定期审查正在进行的临床试验中积累的数据
- 就临床试验参与者的持续安全问题向主办方提供建议
- 就研究的持续有效性和科学价值向发起人提供建议
临床终结点委员会
临床终点委员会(CEC)--也被称为裁决委员会或终点裁决委员会(EAE)--是另一种类型的监督小组,赞助者可以委托其分担临床监测的责任。CECs用于对终点进行集中裁决,以帮助规范结果和优化数据质量。这些类型的委员会在以下情况下是最有价值的。
- 终点是可以解释的,例如,临床事件
- 端点需要应用一个复杂的定义
- 该研究不能是盲目的
- 该研究预计将有较多的入选者或较长的持续时间
- 预计不同研究地点会有全球或文化差异
- 所关注的终点不在研究者的治疗专长范围内
- 需要数据来支持DMC功能或适应性研究设计
CECs通常由一个独立的临床和/或诊断专家小组组成,他们负责。
- 以盲目的、无偏见的、保密的和基于共识的方式对可疑的安全和/或疗效终点进行集中审查和分类
- 确定这些端点是否符合协议的定义
- 为统计分析提供标准化的端点结果
- 将事件分类为与研究设备和/或程序有关(在医疗设备研究中)。
根据FDA指南,CEC应尽可能对治疗进行盲法,即使有关试验没有盲法。1此外,裁定过程的设计应既保持CEC的独立性,又防止任何可能影响其决策的不当偏见。[2]值得注意的是,CEC不进行中期分析或向赞助商提出建议。
比较DMCs和CECs
DMCs和CECs都是由审查研究数据的独立医生组成的。这两种类型的委员会都需要有章程,规定其审查过程和责任,以及没有利益冲突的声明。然而,这些委员会在其目标和结构上有所不同。
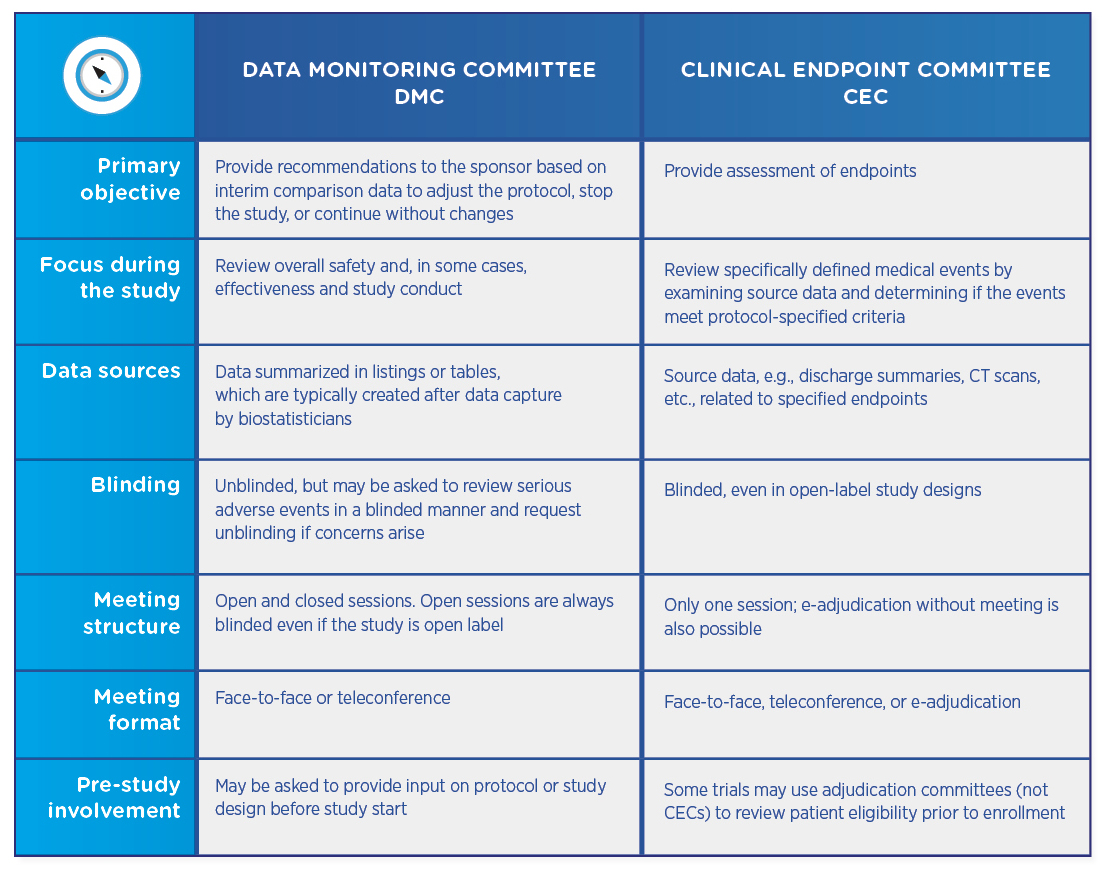
DMC和CEC并不相互排斥,一项审判可以利用其中一个委员会,也可以同时利用两个委员会。如果两个委员会都参与,CEC通常在DMC之前,然后审查已经判决的数据,以确保其尽可能准确和无偏见。
利用DMC或CEC的决定应在临床试验计划过程的早期做出,因为操作和管理这些委员会所涉及的数据交换、协调和沟通会增加复杂性。与了解何时和如何利用这些监督小组的CRO合作伙伴合作,可以帮助申办者以有效的方式管理和运作这些委员会,并根据其临床试验的细微差别而定制。
[1]美国食品和药物管理局。临床试验发起人指南》。临床试验数据监测委员会的建立和运作,2006年3月。可在https://www.fda.gov/downloads/RegulatoryInformation/Guidances/ucm127073.pdf。
[2]Tyner CA, et al. Establishment and Operation of Clinical Endpoint Committees:在整个生物制药行业实施的最佳实践,白皮书。