我们在了解儿科癌症基因组学方面取得了显著进展。 我们对儿科癌症基因组学的了解取得了显著进展,这些进展使我们认识到 这些进步使我们认识到,用于成人癌症适应症研究的产品可能对儿科患者的健康有益。 对儿科患者的健康有益。通过填补孤儿药 通过堵住孤儿药豁免漏洞并尽早与美国食品及药物管理局进行讨论,研究 儿童加速治愈与公平法案》(RACE)有可能加速开发治疗儿童癌症的新方案。 加速儿童癌症治疗新方案的开发。
提交初步儿科研究计划
根据《儿童研究与治疗法案》,分子靶向 儿科癌症分子靶向研究应使用适当的制剂,以获得有关剂量、安全性和 在剂量、安全性和初步疗效方面获得有临床意义的研究数据,为潜在的儿科标签提供信息。 为潜在的儿科标签提供信息[1]。 遵守《儿童研究与治疗法案》的关键第一步是 提交初步儿科研究计划 (iPSP),概述拟议的时间表和儿科研究设计 和儿科研究设计。 研究的拟议时间表和设计。
通常情况下,iPSP 必须在第二阶段结束 (EOP2) 会议后 60 天内提交。 通常情况下,iPSP 必须在第二阶段结束 (EOP2) 会议后 60 天内提交。申办者可考虑在第二阶段研究完成后 在 EOP2 会议之前或会议期间起草 iPSP。 或在 EOP2 会议之前起草 iPSP,以便就成人和儿童研究的终点选择进行战略性讨论。 成人和儿童研究的终点选择进行战略性讨论。如果申办者正在通过 2 期研究寻求加速批准 则 iPSP 应在第一阶段结束(EOP2)后提交。 1 期结束 (EOP1)。如果初始 IND 包括 3 期研究,则 iPSP 应在提交 IND 时提交。
FDA 指南 儿科研究计划:提交初始儿科研究计划和修正初始研究计划的内容和流程[PDF] 概述了 iPSP 的时间安排和预期内容。iPSP 的主要内容包括:[2]
- 儿科疾病概述
- 调查产品概览
- 所有计划进行的非临床和临床研究的表格摘要(见图 1)
- 将研究的所有儿科年龄组的儿科专用配方开发计划详情
- 相关非临床研究数据的简要概述,如果现有数据不充分,则说明将开展的非临床研究
- 支持设计和/或启动儿科研究的临床数据简述
- 计划进行的临床研究概要,包括药代动力学/药效学(PK/PD)研究以及安全性和有效性研究
- 发展时间表
图 1.表格摘要模板示例
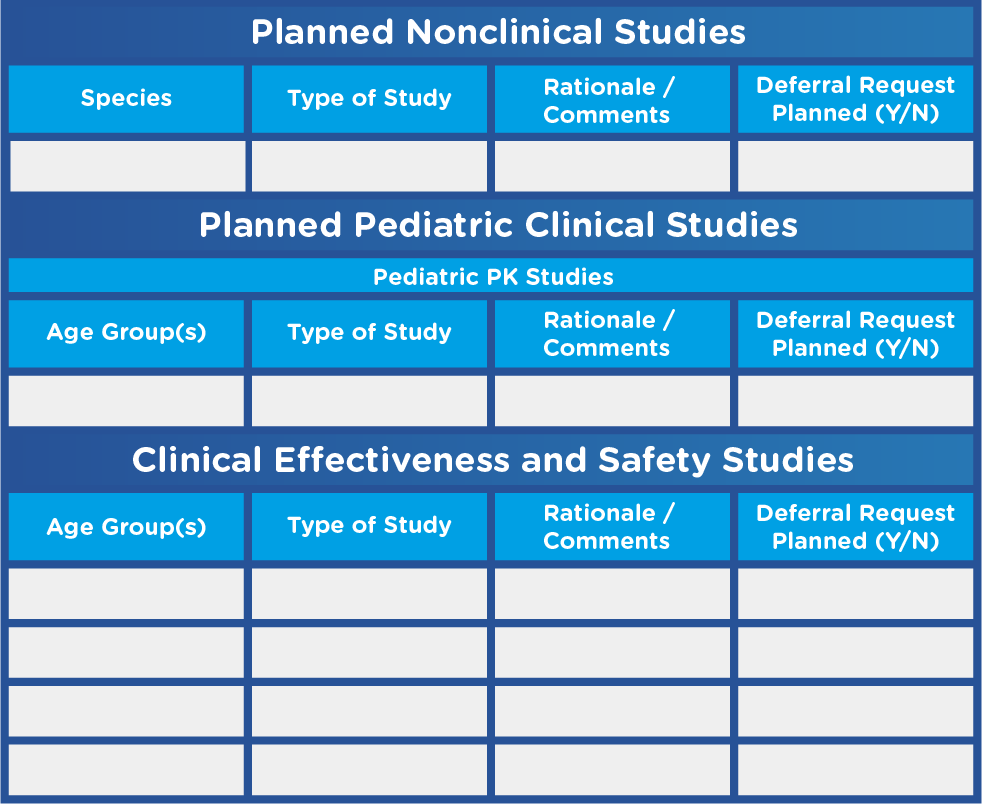
改编自美国食品和药物管理局。儿科 研究计划:提交初始儿科研究计划和修订初始研究计划的内容和程序。 计划和修改后的初始研究计划。
制定 iPSP 时应牢记的关键因素包括
- 需要在与研究产品相关的每个年龄组进行研究
- 不同的儿科年龄组可被视为不同的标签适应症,可能需要不同的产品配方
- 必须精心设计儿科试验,以满足从监管机构和研究人员到儿童及其护理人员等一系列利益相关者的期望
采取行动
虽然《儿童权利法案》使美国对肿瘤产品的儿科要求与欧盟的要求更加一致。 儿科肿瘤产品的要求与欧盟的要求更趋一致,但两者之间仍存在细微但重要的差异。 法规之间存在细微但重要的差异。例如,在欧盟 例如,儿科研究计划 (PIP) 的提交时间不得迟于 除非另有说明。 在欧盟,儿科研究计划 (PIP) 必须在人体药代动力学 (PK) 研究完成后提交,除非另有说明。 此外,PIP 的所有内容都具有约束力,任何修改都必须经 任何修改都必须经过欧洲药品管理局的审查和批准。 此外,向欧洲药品管理局(EMA)提交的新产品/变体或延期的上市许可申请必须包括以下内容 此外,向欧洲药品管理局(EMA)提出的新产品/变体或延期的上市许可申请必须包括验证儿科开发 已按照 PIP 中列出的时间表和计划进行。
规划并最终开展儿科肿瘤试验的需求 肿瘤试验的需求可能令人望而生畏。随着适应症向分子 靶点的转变,申办者面临着重新评估和确定优先次序的挑战。 管线。由于许多产品都有一个共同的分子靶点,临床试验的竞争可能会变得越来越激烈。 竞争会越来越激烈,因为各项研究都在争夺同样少量的患者。 患者。没有儿科经验的专业制药和生物技术公司 可能希望探索行业合作或与合同研究组织(CRO 研究组织 (CRO) 建立合作关系。 和肿瘤学方面具有专长的合同研究组织 (CRO) 建立行业合作或伙伴关系。 了解更多信息,请下载我们的白皮书 白皮书,了解更多信息。 儿科研究的可行性。