随着科学知识、临床经验和对基因治疗产品接受程度的不断发展,确保这些新型治疗方法安全性的监管框架也在不断发展。迄今为止,国际上还没有统一的基因治疗产品监管标准;不过,美国、欧盟和日本已经建立了监管框架,但也有细微的差别。了解基因治疗产品的监管方式--以及如何应对地区或国家监管差异--有助于您制定高效的产品开发计划,为有需要的患者提供治疗。
在这篇博文中,我们将回顾美国、欧盟和日本的基因疗法监管框架,重点介绍旨在简化产品开发和审批流程的计划。
基因治疗监管机构
当监管机构为基因疗法的开发制定了框架时,该框架往往也适用于细胞和组织疗法。鉴于这些框架的范围很广,美国、欧盟和日本的监管机构都成立了专门负责评估基因疗法的委员会或部门:
- 美国- 美国食品和药物管理局及其生物制品评估和研究中心 (CBER) 下设组织和先进疗法办公室。
- 欧盟--除欧洲药品管理局 (EMA) 的人用医药产品委员会外,还有一个专门的先进疗法委员会,负责基因疗法。
- 日本 - 在药品和医疗器械管理局 (PMDA) 以及厚生劳动省之下,设有细胞和组织产品办公室。
这三个国家已建立的基因治疗框架包括促进细胞和基因治疗产品开发、审查和注册的快速计划。
促进产品开发的计划
这些国家都提供有助于简化基因疗法开发的计划。在美国,有三个旨在促进产品开发的计划--快速通道、突破性疗法和再生医学高级疗法(RMAT)指定。由于 "快速通道 "指定是最先开发的,该指定的许多特点也被纳入了 "突破性疗法 "和 "再生医学高级疗法 "指定中。欧盟提供优先药品 (PRIME) 称号,日本提供 Sakigake 称号。
上述所有指定的一个共同点是要求开发中的产品用于治疗与严重发病率相关的严重疾病,而这种疾病的医疗需求尚未得到满足(见图 1)。不同指定的标准措辞各不相同,对未满足医疗需求的要求可间接表述为要求在研产品与现有疗法相比具有更好的疗效或安全性。所有这些称号的另一个共同点是,如果情况发生变化,产品不再符合称号标准,这些称号就会丧失。
值得注意的是,美国的 "快速通道"、欧盟的 "PRIME "和日本的 "Sakigake "允许使用非临床数据,无论是单独使用(快速通道)还是与临床数据结合使用(PRIME 和 Sakigake),以支持指定申请。
图 1.促进基因疗法发展的计划概览
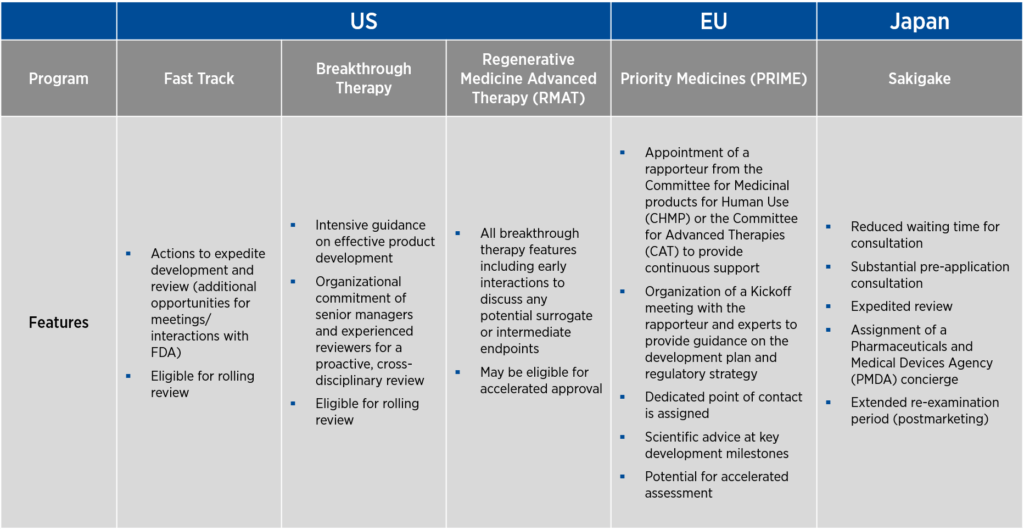
获得这些称号的申办者能更多地接触到授予称号的监管机构,并从监管机构获得高级管理层的反馈意见。通过频繁的会议,申办者和监管机构可以在研究设计和数据要求方面达成一致。
您还将从更大的灵活性中获益,包括在提交生物许可申请 (BLA) 之前将其组成部分提交审查的能力。
加快申请审查的计划
如果产品成功完成临床开发,这三个国家都会提供加快 BLA 或上市申请审查的计划。在美国和日本,这些计划被称为优先审评,而在欧盟,该计划被称为加速评估。与促进产品开发的项目一样,这些加速审评项目也有特定的要求和特点(见图 2)。
图 2.基因治疗快速审查计划概览
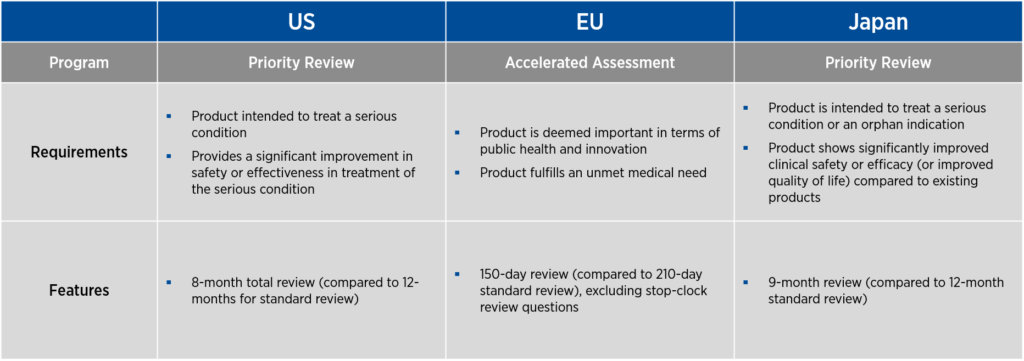
在美国,优先审查的总审查期为 8 个月,而标准审查为 12 个月。在欧盟,加速评估提供 150 天的审查期,不包括停止时钟审查问题,而通常的标准审查期为 210 天。在日本,优先审查的审查期为 9 个月,而标准审查期为 12 个月。
加快注册的计划
有条件批准是一种加快有前景疗法注册途径的机制。每个国家对有条件批准使用不同的术语:
- 美国- 加速审批
- 欧盟- 有条件的上市许可
- 日本- 有条件和有期限的批准
有了这些快速注册途径,您可能只需进行两项关键试验中的第一项,或使用替代终点作为关键研究的疗效终点,即可获得有条件批准。任何快速注册途径的一个关键要素是,在获得有条件批准后,必须及时开展确证研究。
在欧盟和日本,有极少数情况下无需进行确证研究即可获得上市许可。在欧盟,这种情况被称为特殊情况下的上市许可。一旦获得批准,这类上市许可最长可持续五年,但其状态每年会被重新审查一次。在日本,不进行确证研究的有条件批准被称为有条件早期批准制度。
不断变化的景观
在美国,一个相对较新的发展是FDA 的 CBER 产品监管建议初步目标参与会议 (INTERACT),这是一种非正式、无约束力的一小时咨询,使您能够就尚未进入 IND 会议前阶段的问题获得创新研究产品的初步建议。INTERACT 计划适用于 "因使用复杂制造技术、开发创新设备或采用尖端测试方法而导致安全性状况未知,从而带来独特挑战 "1的产品。
有关基因疗法现有监管框架的更多信息,请观看我们的网络研讨会《实现基因疗法的承诺》:克服患者注册和安全挑战的新途径。
主要收获
随着监管机构不断发布新的指南或公布有助于解释法规的资源,申办者必须跟上不断发展的标准和最佳实践。如果您对基因治疗法规或加急项目的资格有任何疑问,请与我们联系,安排对话。
1美国食品和药物管理局。INTERACT 会议。FDA 网站。见https://www.fda.gov/vaccines-blood-biologics/industry-biologics/interact-meetings。2020年10月19日访问。