基因疗法的承诺是提供变革性的治疗方法,切实改善患者的生活质量,目前许多患者都患有使人衰弱的疾病。为了将这些疗法推向市场,申办者的任务是开展临床试验,在适当的安全监督下提供有力的证据。此外,基因治疗试验还面临着无数的后勤障碍,包括寻找合适的患者、将他们与合格的研究机构配对,以及确保他们理解并能满足研究方案的要求。
本文是基因疗法开发博客系列三部曲的第一部分,我们将探讨基因疗法的新兴前景,并讨论研究这些先进疗法的高层次注意事项。
基因治疗的现状
根据美国国立卫生研究院(National Institutes of Health)的定义,罕见病是指在美国患病人数少于 20 万的疾病。据估计,95% 的罕见病目前还没有治疗方法,这就凸显了对基因疗法等创新疗法的需求。
细胞和基因疗法的开发是临床试验活动中一个相对较新且发展迅速的领域,美国食品及药物管理局在 2020 年发布的七份指导文件草案就证明了这一点。然而,尽管有大量候选疗法正在开发中,但进入市场的却寥寥无几,许多障碍依然存在。
基因治疗面临的挑战
基因疗法发展中的障碍包括
- 生产的可扩展性。 目前仍有很大的需求和机会来提高效率和优化生产流程,以经济高效地大规模部署基因疗法。
- 复杂的监管途径。 监管环境不断变化,许多司法管辖区缺乏开发基因治疗产品的既定监管框架。
- 免疫原性问题。 针对基因治疗产品的免疫反应是多因素的,通常需要长期跟踪。
- 剂量确定。 确定安全、有效的剂量可能具有挑战性,因为临床前数据可能无法转化为人体数据,而且高剂量可能会导致严重的健康风险。
基因治疗临床试验的全局考虑
在开发基因疗法和设计临床研究时,申办者应从更高的层面加以考虑:
1.载体类型
治疗遗传疾病最常用的两种病毒类型是慢病毒和腺相关病毒(AAV)。
慢病毒通常用于需要大型囊体的情况。通常,慢病毒载体用于体外基因修饰细胞疗法,即从患者体内获取细胞,对其进行修饰,然后将其送回患者体内。慢病毒将其有效载荷整合到宿主染色体中,因此使用慢病毒载体的疗法需要长达 15 年的长期跟踪。
无论是颅内、眼内、肌内还是静脉注射,AAV 载体通常都是体内给药。使用 AAV 向量的基因疗法通常会成为一个外显子,即细胞核内的环状 DNA 片段,因此需要长达五年的随访。
2.地点和数量
由于患者、关键意见领袖(KOL)和具备开展基因治疗研究必要能力的机构在地理上比较分散,申办者需要确定在哪里开展研究以及启动多少个研究点。关键在于确定哪些主要研究者曾在感兴趣的患者群体中开展过研究,以及他们是否隶属于能够开展研究的机构。
3.评估类型
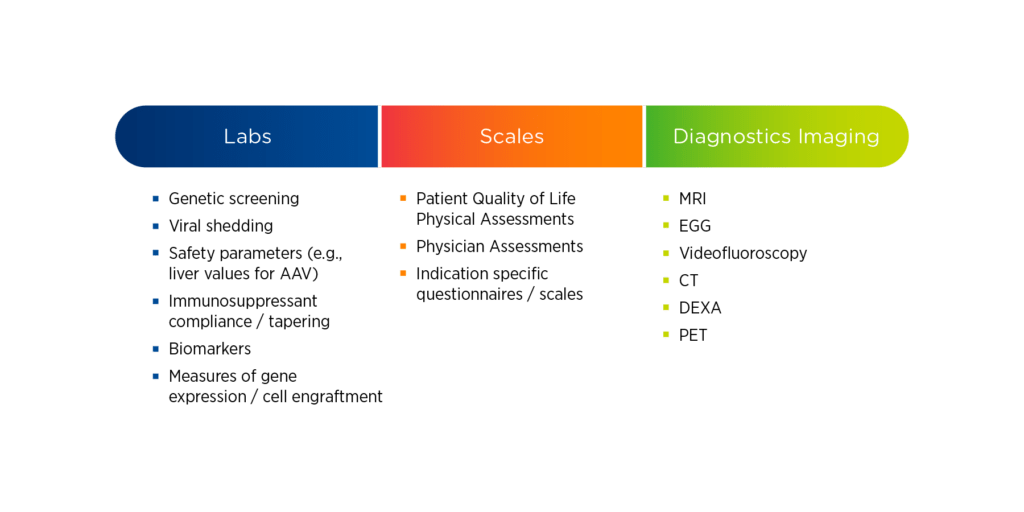
基因治疗研究往往是资源密集型的,需要研究机构进行各种评估。这些评估可能包括简单和复杂的实验室测试、量表和评估以及特殊成像。对于每项评估,都必须了解谁有资格执行、需要执行的频率以及在哪里执行。
对于脆弱的儿科人群来说,实验室评估可能比较复杂,因此可能需要专门的供应商。对于量表和评估,申办者应考虑评分者是否需要特殊资格、如何处理评分者之间的可靠性,以及这些评估是否可以通过家庭医疗或移动医疗服务进行。对于特殊成像要求,可能需要特定的校准、协议和验证;这可能会限制成像的执行地点。
图 1.基因治疗研究中的常见评估
在本系列博客的第二部分中,我们将探讨基因治疗试验设计的关键注意事项。
在 Premier,我们在过去五年中开展了 60 多项细胞和基因治疗研究。我们在病毒载体方面拥有丰富的经验,在肿瘤学、罕见病和儿科研究方面拥有深厚的治疗专业知识。单击此处了解更多有关 Premier 如何帮助推进基因治疗研究的信息。